Фенилкетонурия - класически признаци, като наследствена и диетична терапия
Съдържание
- 1Как се проявява болестта на фенилкетонурията
- 2Механизъм на развитие на болестта
- 3Фенилкетонурия при деца
- 4Симптоми на заболяването
- 5Причини и провокативни фактори
- 6Диагностика
- 7Лечение на класическа фенилкетонурия
- 8Хранене на новородени и терапия с диета
- 9Диета за деца в предучилищна възраст и ученици
- 10Групи продукти на ФКУ
- 11Как се контролира нивото на фенилаланин в кръвта
- 12Видео
Заболяването, чието появяване е свързано с дефекти в генетичния клетъчен апарат - фенилкетонурия - е включено в малък списък от наследствени заболявания, които се лекуват. Откривателят на тази болест беше норвежкият лекар IA Felling, по-късно беше открито, че развитието и протичането на заболяването съответства на един ген, наречен фенилаланин хидроксилазен ген (дълъг рамо на 12-та хромозома, съдържащ до 4,5% от целия ДНК клетъчен материал). Наследственият дефект води до частична или пълна дезактивация на ензима на черния дроб от фенилаланин-4-хидроксилаза.
Как се проявява болестта на фенилкетонурията
Наследствено заболяване на фенилкетонурия (PKU) води до хронично отравяне на организма с токсични вещества, образувани поради нарушен метаболизъм на аминокиселините и процесахидроксилиране на фенилаланин. Постоянната интоксикация причинява увреждане на централната нервна система (ЦНС), чиято проява е прогресивното намаляване на интелигентността (фенилпирувинова олигофрения).
Болестта на изсичането се проявява в прекомерно натрупване на фенилаланин в организма и в продуктите на неговия неподходящ метаболизъм. Други фактори за развитие на фенилкетонурия включват счупен транспорт на аминокиселини през кръвно-мозъчната бариера, ниски нива на невротрансмитери (серотонин, хистамин, допамин). При липса на навременно лечение на заболяването води до умствена изостаналост и може да причини смъртта на детето.
Механизъм на развитие на болестта
Инхибирането на метаболита, причинено от мутационното инактивиране на ензима, е активирането на спомагателните пътища за обмен на фенилаланин. Ароматната алфа-аминокиселина, в резултат на дефектни обменни процеси, се разпада на токсични производни, които не се образуват при нормални условия:
- фенилпирувинова киселина (фенилпируват) - мастен ароматен алфа-кетоацид, неговото образованиеводи до миелинизация на процесите на неврони и деменция;
- фенилмаксична киселина - продукт, образуван по време на регенерацията на фенил вирионова киселина;
- Фенилетиламин - първоначално съединение за биологично активни предаватели на електрохимични импулси, увеличава концентрацията на допамин, адреналин и норепинефрин;
- Ортофенилацетат - токсично вещество, което причинява нарушаване на метаболитните процеси на фекалните съединения в мозъка.
Медицинските статистически данни показват, че патологично изменен ген присъства в 2% от населението, но не се проявява. Генетичен дефект се предава на детето от родителите само при наличието на заболяването при двамата партньори, като при 50% от случаите бебето става носител на мутиралия ген, оставайки здрав. Вероятността фенилкетонурия при новородени да доведе до заболяването е 25%.
С какъв тип се наследява
Болестта на изсичане е генетично отклонение, отпечатано върху автозомно-рецесивен тип. Този тип наследяване означава, че развитието на признаците на вродено заболяване се случва само когато едно дете е наследено от дефектен генетичен екземпляр на двамата родители, които са хетерозиготни носители на променения ген.
Развитието на вродени заболявания в 99% от случаите причинява мутация на гена, отговорен за ензимното кодиране, който осигурява синтеза на фенилаланин-4-хидроксилаза (класическа фенилкетонурия). До 1% от генетичните заболявания са свързани с мутационни промени, настъпващи в други причиняващи генинедостатъчност на дихидроптеридин редуктаза (тип II PKU) или тетрахидробиоптерин (PKU тип III).Фенилкетонурия при деца
PKU тип II се характеризира с факта, че първите клинични симптоми се появяват след 1,5 години от момента на настъпване на светлината. Признаците на болестта не изчезват след диагностициране на генетични аномалии и започване на диетична терапия. Този тип вродено заболяване често води до фатален изход от 2-3 години от живота на детето. Най-честите симптоми на PKU тип II са:
- изразени отклонения в умственото развитие;
- хиперрефлексия;
- нарушение на двигателните функции на всички крайници;
- синдром на неконтролирани мускулни контракции.
Клиничните признаци на мутационни промени в гените от тип III са подобни на болестите, възникващи при тип II. Дефицитът на тетрахидробиоптерин се характеризира с триада от специфични симптоми:
- висока степен на умствена изостаналост;
- очевидно намален размер на черепа спрямо други части на тялото;
- спастичност на мускулите (с възможност за пълна загуба на движение на крайниците).

Прояви на болестта на изсичането
В хода на клиничните проучвания и наблюденията се предполага, че ефектът е билтоксични производни на обмена на фенилаланин причинява намаляване на интелектуалните способности, което е прогресивно и може да доведе до деменция (олигофрения, идиоти). Сред вероятните причини за необратими нарушения на мозъчната активност, най-разумно се счита, че се дължи на намаляване на нивата на тирозин, липсата на невротрансмитери, които предават импулси между невроните.
Точната причинно-следствена връзка между наследствените заболявания и мозъчните нарушения не е разкрита досега, както и механизмът на развитие, дължащ се на фенилкетонурия на такива психични състояния като епраксия, ехолалия, свирепи атаки и раздразнителност. Резултатите от анализите показват, че фенилаланинът има директен токсичен ефект върху мозъка, което също може да доведе до намаляване на интелигентността.
Структурни и фенотипни особености
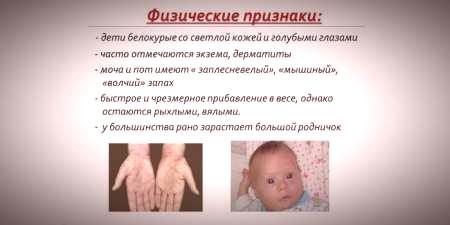
Като се има предвид, че наситеността на пигмента на кожата и косата зависи от нивото на тирозин в митохондриите на хепатоцитите и фенилкетонурията води до спиране на превръщането на фенилаланин, пациентите с това заболяване имат фенотипни признаци (рецесивни признаци). Повишен мускулен тонус причинява появата на отклонения в структурата на тялото - става диспластичен. Отличните външни характеристики на фенилкетонурията включват:
- хипопигментация - светла кожа, бледосини очи, обезцветена коса;
- цинизъм на крайниците;
- намален размер на главата;
- специфичното положение на тялото - когато се опитвате да стоите или седите детето приема позицията на "шивач" (ръцете и краката са свити в ставите).
Симптомиболест
Навремето, болестта на Felling се подлага на успешно лечение чрез регулиране на храненето и развитието на детето се извършва в съответствие с възрастовата му група. Трудността при откриването на генна мутация се крие във факта, че ранните признаци са трудни за откриване дори на опитен педиатър. Тежестта на симптомите на вродени заболявания нараства с нарастването на детето, тъй като употребата на протеинови храни допринася за развитието на разстройства на ЦНС.
Признаци при новородени
През първите дни от живота на детето е трудно да се открият признаци на патологични аномалии - бебето се държи естествено, няма забавяне в развитието. Симптомите на заболяването първо се появяват след 2-6 месеца след раждането. Родителите трябва да бъдат нащрек за поведението на бебето, което се характеризира с ниска активност, летаргия или, обратно, тревожност, свръхчувствителност.
С началото на кърменето, новите протеини започват да влизат в тялото на новороденото с мляко, което служи като катализатор за появата на първите признаци, което ясно показва, че болестта започва да се развива. Специфичните клинични прояви на заболяването включват:
- постоянно повръщане (често се приема за вродено стеснение на вратаря);
- честа дислокация;
- няма реакция към външни стимули;
- мускулна дистония (намалено мускулно напрежение);
- конвулсивен синдром (припадъци от епилептичен или неепилептичен тип).
Симптоми при деца след 6 месеца
Ако проявата на генетичното заболяване не е такаванастъпили (или не бяха наблюдавани) през първите 6 месеца от раждането на детето, след това след този период вече може точно да се определи изоставането в психомоторното развитие. Симптомите на генетични заболявания, причинени от дефицит на ензими при деца на възраст над шест месеца, са:
- намаляване на активността (до пълно безразличие);
- липса на опити да се изправи, седнал;
- специален "миши" аромат на кожата (миризмата на мухъл възниква поради оттеглянето на токсични фенилаланинови производни чрез потни жлези и урина);
- загуба на способността да се визуализират лицата на родителите;
- лющене на кожата;
- появата на дерматит, екзема, склеродермия.
Прогресия на заболяването при липса на лечение в детска възраст
Ако в детството не са открити отклонения в развитието и не е проведено подходящо лечение, болестта започва да напредва активно и често води до увреждане. Липсата на терапия в ранен стадий на заболяването причинява появата на такива симптоми на заболяването на възраст от 1,5 години:
- микроцефалия (намален размер на мозъка);
- отток (изместване на горната част на зъба напред);
- по-късно изригване на зъбите;
- хипоплазия на емайла (тънка или пълна липса на зъбен емайл);
- забавяне на езиковото развитие до пълната липса на реч;
- 3, 4 степен на олигофрения (умствено изоставане, умствено изоставане);
- вродено сърдечно заболяване (дефекти в структурата на сърдечния мускул, сърцето,големи съдове);
- нарушения на вегетативната система (акроцианоза, повишено изпотяване, артериална хипотония);
- запек.
Причини и провокативни фактори
За да се прояви мутация с автозомно рецесивно наследяване, дефектен ген трябва да се наследи от двамата родители. Генетичните заболявания от този тип се срещат с една и съща честота при новородените момчета и момичета. Патогенезата на ФКУ се причинява от нарушение на обмена на фенилаланин, което може да се появи в 3 форми. Лечението с диетична терапия е предмет на класическа фенилкетонурия от тип I. \ t
Атипичните форми на болестта могат да бъдат излекувани чрез регулиране на храненето. Тези отклонения се дължат на дефицита на тетрахидроптерин, дехидроптерин редуктаза (рядко - пируватетрахидроптеринстанца, гуанозин-5-трифосфат циклохидролаза и др.). Повечето случаи на фатални случаи са регистрирани при пациенти с редки вариации на PKU, като клиничните прояви на всички форми на заболяването са сходни. Рискът от раждане с мутирал ген на фенилаланин хидроксилазата се увеличава, ако родителите са близки роднини (с близки бракове).
Диагностика
В случай на съмнение за генетично заболяване диагнозата се установява въз основа на съвкупността от данни, получени в резултат на изследването на анамнезата на заболяването - генеалогични данни, резултатите от клиничните и медицинските генетични изследвания. За навременно откриване на вродени заболявания (PKU, кистозна фиброза, галактоземия и др.) Програма за задължителна масаЛабораторно изследване на всички новородени (неонатален скрининг).
Ако бъдещите родители са наясно с наличието на мутирал ген, съвременната медицина предлага начини за откриване на дефект по време на бременността (пренатална диагностика на плода чрез инвазивен метод). За разделяне на фенилкетонурия по вид на тежест се използва условна класификация въз основа на нивото на фенилаланин в фибриногенната течност, получена от плазмената плазма:
- Тежка фенилкетонурия - 1200 mkmol /l.
- Средно - 60-1200 μmol /l.
- Лесно (не се изисква лечение) - 480 μmol /l.
Скринингов тест
Откриването на генетични аномалии се наблюдава в няколко етапа. На първия етап в болницата при всички бебета се извършва 3-5 дни живот, като се вземат проби от периферна кръв (от пет) за изследване. Материалът се полага върху хартиен носител и се изпраща в биохимична лаборатория, където се провежда биохимичният му анализ. На втория етап на скрининговия тест се определя концентрацията на фенилаланин с нормална стойност.

Ако не бъдат открити патологични промени, диагнозата приключва, което се записва в картата на детето. При наличие на отклонения от нормата, резултатите от диагнозата се изпращат на лекар-педиатър, за да се осигури прецизно изследване на кръвна проба от новородено. Здравето на детето зависи от навременното и точно изпълнение на всички мерки за откриване на аномалии. Ако диагнозата се потвърди след повторен скринингов тест, родителите на детето ще го направятизпратени в клиниката за детска генетика с цел лечение.
Анализи и проучвания за потвърждаване на диагноза
Повторното диагностициране при откриване на отклонение от нормата по време на първоначалния скринингов тест се извършва чрез преоценка на анализите. В допълнение към определянето на съдържанието на фенилаланин в кръвта към методите за диагностика на ФКУ при деца и възрастни включват:
- Тест за разрушаване - определяне на фенилпирувинова киселина в урината чрез добавяне на железен хлорид към биоматериала (има синьо-зелено оцветяване);
- Gatree тест - оценка на степента на реакция на микроорганизмите към метаболитни продукти или ензими, съдържащи се в кръвта на пациента;
- хроматография - изследване на химичните свойства на веществата, разпределени между две фази;
- флуориметрия - облъчване на биоматериал чрез монохроматично лъчение, за да се определи концентрацията на съдържащите се вещества;
- електроенцефалография - диагностика на електрическата активност на мозъка;
- магнитен резонанс - нарушаване на атомните ядра на клетките чрез електромагнитни вълни и измерване на техния отговор.
Лечение на класическа фенилкетонурия
Основата на терапията с фенилкетонурия е ограничаването на консумацията на продукти, което е източник на протеини от животински и растителен произход. Единственият начин за успешно лечение е диетична терапия, адекватността на която се оценява от съдържанието на фенилаланин в серума. Максимално допустимо ниво на аминокиселини при пациенти от различни възрастови групие:
- при новородени и деца под 3 години - до 242 μmol /l;
- за деца в предучилищна възраст - до 360 mkmol /l;
- при пациенти на възраст от 7 до 14 години - до 480 μmol /L;
- при юноши - до 600 μmol /l.
Ефектът от диетата зависи от това кой етап от заболяването е корекцията на диетата. При ранна диагностика на вродена патология се предписва диетична терапия от 8 седмици от живота (след този период вече започват необратими промени). Липсата на своевременни мерки води до усложнения и намаляване на нивото на интелигентност с 4 пункта след 1 месец от раждането до началото на лечението.

Като се има предвид, че терапевтичната диета за фенилкетонурия включва пълно изключване от диетата на животинските протеини, е необходимо да се използват други източници на незаменими аминокиселини, както и витамини от група В, съдържащи калций и фосфор минерални съединения. Продуктите, предназначени за добавяне към не-протеиновата диета, включват:
- протеинови хидролизати (амигени, аминазол, фибринози);
- не съдържат смеси на фенилаланин, наситени с незаменими аминокиселини - тетрафен, без фенил.
Наред с лечебните мерки за елиминиране на причината за функционирането на организма, трябва да се извърши симптоматично лечение, насочено към елиминиране на дефекти в речта и нормализиране на координацията на движенията. Комплексната терапия включва физиотерапевтични процедури, масаж, помощ от логопед, психолог, изпълнение на гимнастически упражнения. В някои случаи с диетична терапияпоказва използването на антиконвулсанти, ноотропни и съдови медикаменти.
Особености на третирането на атипични форми
Фенилкетонурия тип II и III не може да се лекува с диета с ниско съдържание на протеини - нивото на фенилаланин в кръвта остава непроменено, за да се ограничи притока на протеин в организма, или клиничните симптоми напредват, дори когато нивото на аминокиселината се понижи. Ефективната терапия на тези форми на заболяването се извършва с:
- тетрахидробиоптерин - фактор на засегнатия ензим;
- синтетични аналози на тетрахидробиоптерин - тези вещества са по-добре проникнали през кръвно-мозъчната бариера;
- лекарства за заместителна терапия - не елиминират причината за фенилкетонурия, но подкрепят нормалното функциониране на организма (леводопа заедно с карбидопа, 5-окситриптофан, 5-формилтетрахидрофолат);
- хепатопротектори - подпомагат функционирането на черния дроб;
- антиконвулсанти;
- въвеждане на гена на фенилаланин хидроксилаза в черния дроб - експериментален метод.
Характеристики на храненето на новородените и терапия за диета
През първата година от живота на детето е допустимо да се кърми, но количеството му трябва да бъде ограничено. До 6 месеца приемливото ниво на консумация на фенилаланин е 60-90 mg на 1 kg тегло на бебето (в 100 g мляко съдържа 5,6 mg фенилаланин). От 3 месеца диетата на бебето трябва постепенно да се разширява чрез въвеждане на плодови сокове и картофено пюре.
За деца от 6 месеца е позволено въвеждането в растителна растителна пюре, каша (от саго), без протеиниКиселев. След 7 месеца можете да дадете на бебето ниско-протеинови макаронени изделия, с 8 месеца - хляб без протеини. Не е установена възрастта, в която трябва да се ограничи предаването на протеин в тялото на болно дете. Лекарите все още обсъждат въпроса за целесъобразността на диетичната терапия, но са съгласни, че най-малко 18-годишна възраст, диетичното хранене трябва да се спазва.
Фенилкетонурия, диагностицирана с жена, не е причина да се откаже от раждането на дете. Бъдещи майки с ФКУ за предотвратяване на увреждане на плода по време на бременност и за предотвратяване на възможни усложнения са необходими преди планираното зачеване и по време на носене на детето да се придържа към диетата с ограничаване на фенилаланин (нивото в кръвта му трябва да бъде до 242 микромола на литър).
Смеси за ваксини за бебета
Диета с фенилкетонурия се основава на значително намаляване на дозата на естествения протеин в дневния хранителен режим, но тялото на новороденото бебе не може да се развие нормално при липсата на необходимите микроелементи. За да се запълни нуждата на бебето от протеини, се използват антибактериални аминокиселини, които според руското законодателство трябва да се предоставят безплатно.
Толерантността на детето към фенилаланин през първата година от живота бързо се променя, така че е необходимо да се контролира концентрацията му в кръвта на бебето и да се направят корекции в диетата. Смесите са за определени възрастови групи:
- за бебета до годината, назначен за Афенилак 15, Аналогов JV, PKU-1, PKU-mix, PKU Anamix;
- за деца над 1 годинагодина, предписват обогатени с витамини и минерали смес от високо съдържание на протеини - PKU Prima, P-AM Universal, PKU-1, PKU-2, HRM Maxamed, HR Maxumum.

Диетични продукти за попълване на протеинови запаси
Един от основните компоненти на диетичната диета за фенилкетонурия е храни с ниско съдържание на протеини на базата на нишесте. Тези добавки съдържат хидролизат казеин, триптофан, тирозин, метионин, азот и осигуряват ежедневната нужда на детето от протеин на детето, което е от съществено значение за нормалното развитие и растеж. Специализирани продукти, които запълват липсата на необходимите минерали и аминокиселини в липсата на диета, са:
- Berlofen;
- Tsimorgan;
- Minafen;
- Апотони.
Диета за деца в предучилищна възраст и ученици
Тъй като тялото се адаптира към фенилаланин при деца на възраст над 5 години, е възможно постепенно да се намалят диетичните ограничения. Разширяването на диетата е чрез въвеждане на зърнени храни, млечни продукти, месни продукти. Учениците в гимназията вече имат висока толерантност към фенилаланин, така че в тази възраст можете да продължите да разширявате диетата, докато е необходимо да следите отговора на всички промени в диетата. За наблюдение на състоянието на детето се използват следните методи:
- оценка на неврологичните параметри, психологичното състояние;
- контрол на параметрите на електроенцефалограмата;
- определяне на нивото на фенилаланин.
Продуктови групи на PKU
В диетата на пациенти с PKU заедно с не-протеиниНишестените продукти и терапевтичните смеси включват също продукти от естествен произход. При съставянето на менюто трябва ясно да се изчисли количеството на консумираните протеини и да не се превишава препоръчаната дозировка от лекар. За да се изключат токсичните ефекти върху тялото, се разработват 3 списъка с продукти, които съдържат забранени (червени), непредставени (оранжеви) и разрешени (зелени) позиции.
Червен списък
Фенилкетонурия се развива на фона на отсъствието на ензим, който се превръща в тирозин фенилаланин, така че високото съдържание на протеини е основа за присвояване на продукти в забранения (червен) списък. Позициите от този списък трябва изцяло да изключват диетата на пациента на PKU:
- месо;
- вътрешни органи на животни, странични продукти;
- колбаси, колбаси;
- морски храни (включително риба);
- яйца от всички птици;
- кисели млечни продукти;
- гайки;
- бобови и зърнени култури;
- соеви продукти;
- желатинови съдове;
- сладкарски изделия;
- аспартам.
Оранжев списък
Продуктите, които се вкарват в тялото на дете, диагностицирано с PKU, са включени в оранжевия списък. Включването в хранителния режим на артикули от този списък е допустимо, но в строго ограничени количества. Въпреки че тези продукти не съдържат много протеини, но мога също да повишавам нивото на фенилаланин, затова не се препоръчва тяхното използване:
- консервирани зеленчуци;
- ястия от картофи и ориз;
- зеле;
- мляко;
- шербет.
Зелен списък
Не-протеинови продукти са разрешени за употреба при пациенти с фенилкетонурия без никакви ограничения. Преди да закупите продукти от зеления списък, трябва да разгледате състава, посочен върху опаковката, и да се уверите, че няма фенилаланин, съдържащ багрило на аспартам:
- плодове;
- зеленчуци (с изключение на картофи и зеле);
- плодове;
- Зелените;
- нишестени зърнени култури (саго);
- мед, захар, конфитюр;
- Брашно от брашно от царевица или ориз;
- масло, мазнини (масло, слънчоглед, маслини).
Как да се контролира нивото на фенилаланин в кръвта
Фенилкетонурията е неизлечима болест, която може да се трансформира във фаза на стагнация чрез прилагане на диетична терапия и терапевтични и профилактични мерки. При промяна на условията на живот, нарушение на диетата, заболяването може отново да се влоши, така че пациентите се нуждаят от наблюдение през целия живот. Процесът на контрол е да се определя периодично нивото на фенилаланин в кръвта. Честотата на доставяне зависи от възрастта на пациента:
- до 3 месеца - скрининг на кръв трябва да се прави веднъж седмично, докато се получат стабилни резултати;
- от 3 месеца до 1 година - 1-2 пъти месечно;
- от 1 до 3 години - 1 път на 2 месеца;
- над 3 години - тримесечно.

Кръвта за анализи изглежда е 3-4 часа след хранене. В допълнение към скрининга, развитието на PKU се контролира чрез определяне на хранителния статус, физическото, емоционалното развитие на пациента, нивото на интелектуалните способности.и развитие на езика. Според резултатите от наблюденията може да е необходима допълнителна диагностика с участието на съответните специалисти.
Видеоклипове
Информацията, предоставена в статията, е информативна. Материалите на статията не изискват независимо третиране. Само квалифициран лекар може да диагностицира и да дава съвети за лечението въз основа на индивидуалните характеристики на конкретен пациент.








